LUYOR-3109高强度紫外催化光源促销
LUYOR-3109紫外光源采用了9颗365nm大功率led,安装有二次光学透镜,输出紫外线强度高,...
2024-08-08

作者:生命科学事业部时间:2019-11-17 19:35:26浏览15755 次
过表达载体主要是将目的基因的编码区(CDS)构建到相应的质粒或者病毒载体中,达到在目的基因过量表达的作用。常规过表达载体主要元件:Promoter—gene—linker—Fluorescent gene—抗药筛选gene。基因过表达载体又可以分为广泛启动子基因表达载体,组织特异性启动子基因表达载体和诱导启动子基因表达载体等,以达到不同研究所需要的目的。
过表达载体主要是将目的基因的编码区(CDS)构建到相应的质粒或者病毒载体中,达到在目的基因过量表达的作用。常规过表达载体</B>主要元件:Promoter—gene—linker—Fluorescent gene—抗药筛选gene。基因过表达载体又可以分为广泛启动子基因表达载体,组织特异性启动子基因表达载体和诱导启动子基因表达载体等,以达到不同研究所需要的目的。
载体构建(vectorconstruction):为把DNA分子运送到受体细胞中去,必须寻找一种能进入细胞、在装载了外来的DNA片段后仍能照样复制的运载体。理想的运载体是质粒(plasmid),在基因工程中,常用人工构建的质粒作为载体。
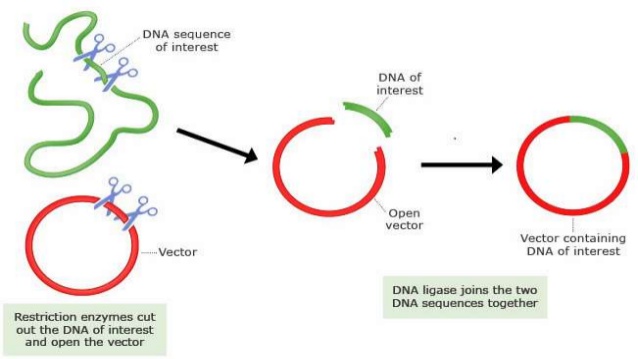
过表达载体的构建方法及步骤
一、载体的选择及如何阅读质粒图谱
目前,载体主要有病毒和非病毒两大类,其中质粒 DNA 是一种新的非病毒转基因载体。
一个合格质粒的组成要素:
(1)复制起始位点 Ori 即控制复制起始的位点。原核生物 DNA 分子中只有一个复制起始点。而真核生物 DNA 分子有多个复制起始位点。
(2)抗生素抗性基因 可以便于加以检测,如 Amp+ ,Kan+
(3)多克隆位点 MCS 克隆携带外源基因片段
(4) P/E 启动子/增强子
(5)Terms 终止信号
(6)加 poly(A)信号 可以起到稳定 mRNA 作用
选择载体主要依据构建的目的,同时要考虑载体中应有合适的限制酶切位点。如果构建的目的是要表达一个特定的基因,则要选择合适的表达载体。

过表达实例--pCAGGS质粒转染小鼠原代神经元
载体选择主要考虑下述3点:
【1】构建 DNA 重组体的目的,克隆扩增/基因表达,选择合适的克隆载体/表达载体。
【2】.载体的类型:
(1)克隆载体的克隆能力-据克隆片段大小(大选大,小选小 ) 。如<10kb 选质粒。
(2)表达载体据受体细胞类型-原核/真核/穿梭,E.coli/哺乳类细胞表达载体。
(3) 对原核表达载体应该注意: 选择合适的启动子及相应的受体菌, 用于表达真核蛋白质时注意克服4个困难和阅读框错位;表达天然蛋白质或融合蛋白作为相应载体的参考。
【3】载体 MCS 中的酶切位点数与组成方向因载体不同而异,适应目的基因与载体易于链接 , 不能产生阅读框架错位。
综上所述,选用质粒(最常用)做载体的5点要求:
(1)选分子量小的质粒,即小载体(1-1.5kb)→不易损坏,在细菌里面拷贝数也多(也有大载体) ;
(2)一般使用松弛型质粒在细菌里扩增不受约束,一般 10个以上的拷贝,而严谨型质粒<10个。
(3)必需具备一个以上的酶切位点,有选择的余地;
(5)满足自己的实验需求,是否需要包装病毒,是否需要加入荧光标记,是否需要加入标签蛋白,是否需要真核抗性(如Puro、G418)等等。
无论选用哪种载体, 首先都要获得载体分子, 然后采用适当的限制酶将载体 DNA 进行切割, 获得线性载体分子,以便于与目的基因片段进行连接。
如何阅读质粒图谱
步:首先看 Ori 的位置,了解质粒的类型(原核/真核/穿梭质粒)
第二步:再看筛选标记,如抗性,决定使用什么筛选标记。
(1)Ampr 水解β-内酰胺环,解除氨苄的毒性。
(2)tetr 可以阻止四环素进入细胞。
(3)camr 生成氯霉素羟乙酰基衍生物,使之失去毒性。
(4)neor(kanr) 氨基糖苷磷酸转移酶 使 G418(长那霉素衍生物)失活
(5)hygr 使潮霉素β失活。
第三步:看多克隆位点(MCS) 。它具有多个限制酶的单一切点。便于外源基因的插入。如果在这些酶切位点以外有外源基因的插入,会导致某种标志基因的失活,而便于筛选。决定能不能放目的基因以及如何放置目的基因。
第四步:再看外源 DNA 插入片段大小。质粒一般只能容纳小于10Kb 的外源 DNA 片段。一般来说,外源 DNA 片段越长,越难插入,越不稳定,转化效率越低。
第五步:是否含有表达系统元件,即启动子-核糖体结合位点-克隆位点-转录终止信号。这是用来区别克隆载体与表达载体。克隆载体中加入一些与表达调控有关的元件即成为表达载体。选用那种载体,还是要以实验目的为准绳。
第六步:启动子-核糖体结合位点-克隆位点-转录终止信号
(1)启动子-促进 DNA 转录的 DNA 序列,这个 DNA 区域常在基因或操纵子编码序列的上游,是 DNA 分子上可以与 RNApol 特异性结合并使之开始转录的部位,但启动子本身不被转录。
(2) 增强子/沉默子-为真核基因组(包括真核病毒基因组)中的一种具有增强邻近基因转录过程的调控顺序。其作用与增强子所在的位置或方向无关。即在所调控基因上游或下游均可发挥作用。/沉默子-负增强子,负调控序列。
(3)核糖体结合位点/起始密码/SD 序列(Rbs/AGU/SDs) :mRNA 有核糖体的两个结合位点,对于原核而言是 AUG(起始密码)和 SD 序列。
(4) 转录终止序列(终止子)/翻译终止密码子:结构基因的最后一个外显子中有一个 AATAAA的保守序列,此位点 down-stream 有一段 GT 或 T 富丰区,这2部分共同构成 poly(A)加尾信号。结构基因的最后一个外显子中有一个 AATAAA 的保守序列,此位点 down-stream 有一段GT 或 T 富丰区,这2部分共同构成 poly(A)加尾信号。
质粒图谱上有的箭头顺时针有的箭头逆时针, 那其实是代表两条 DNA 链, 即质粒是环状双链DNA,它的启动子等在其中一条链上,而它的抗性基因在另一条链上 .根据表达宿主不同,构建时所选择的载体也会不同。
二、目的基因的获得
一般来说,目的基因的获得有三种途径:
调取基因:根据目的基因的序列,设计引物从含有目的基因的cDNA中通过PCR的方法调取目的基因,链接到克隆载体挑取单克隆进行测序,以获得想要的基因片段,这种方法相对成本较低,但是调取到的基因往往含有突变,还有不同基因的表达丰度不同,转录本比较复杂,或是基因片段很长,这些情况都很难调取到目的基因。
全基因合成:根据目的基因的DNA序列,直接设计合成目的基因。此方法准确性高,相对成本会高一些,个人操作比较困难,需要专业的合成公司完成。优点是可以合成难调取及人工改造的任何基因序列,同时可以进行密码子优化,提高目的基因在宿主内的表达量。
三、克隆构建
目前,克隆构建的方法多种多样,除了应用广泛的酶切链接以外,现在还有很多不依赖酶切位点的克隆构建方式。下面具体说一下双酶切方法构建载体的步骤。
(2)X基因慢病毒载体的构建
X基因 基因由Transheep全基因合成,构建于载体PUC57中。PUC57-X基因 EcoRI/BamHI酶切结果:
Lane1:GeneRay 1kb DNA Ladder (从上至下依次为:3000bp, 2000bp, 1500bp, 1000bp, 500bp, 250bp,100bp)
Lane2:PUC57-Neurod1 EcoRI/BamHI酶切产物
酶切完成后进行胶回收
2. 载体用pCDNA3.1双酶切,酶切体系如下。
20ul酶切体系 37度3小时
酶切完成后胶回收(见附录)
Lane1:GeneRay 1kb DNA Ladder (从上至下依次为:12000bp, 8000bp, 6000bp, 5000bp, 4000bp, 3000bp, 2500bp, 2000bp, 1500bp, 1000bp, 750bp, 500bp, 250bp)
Lane2: pcDNA3.1载体酶切回收产物
转化 (感受态细胞: DH5a),具体步骤见附录转化部分。
抗性: Amp; 37℃,培养过夜
转化后X基因分别平板挑菌, 37℃ 250转/分钟摇菌14小时,PCR鉴定后,将阳性菌液送上海权阳生物技术有限公司测序。
X基因 慢病毒载体单克隆菌落PCR鉴定(使用载体通用引物,PCR条带大小应比实际大200bp左右):
Lane1:GeneRay 1kb DNA Ladder (从上至下依次为:2000bp, 1500bp, 1000bp, 750bp, 500bp, 250bp,100bp)
Lane2-4: X基因 菌落鉴定PCR产物
载体构建中需要注意些什么?
一、PCR 过程中的经验教训
做分子克隆,构建表达载体的过程中,目的片段必须跟genebank 中的序列完全一致才行,所以在 PCR 过程中酶的选择首先应选择高保真酶,尤其是当目的片段较长的时候更是如此。这样才能在 PCR 过程中减少错配情况。高保真酶常常对退火温度有要求,对一般的酶来说,退火温度是 Tm-5 度,但如 NEB 的高保真酶的退火温度是 Tm 值+3 度。PCR 循环数不宜太大,20-30 即可。我就曾经用普通的酶进行过 PCR,扩增时出现非特异条带,T-A 克隆后送去测序,发现碱基有错配,而且每次都不一样。
二、PCR 产物直接酶切后连接不成功
当你获得 PCR 产物后,首先是进行纯化、酶切、纯化,然后与酶切后胶回收的载体连接,如果连接成功,当然是恭喜了。但往往会出现连接不成功的,原因以 PCR 产物没有酶切成功可能性大,这可能与引物设计的好坏有关。因为这个时候 PCR 产物是否酶切成功是无法鉴定的,所以你反复尝试几次还不成功的话,就赶紧做个 T-A 克隆吧
三、T-A 克隆应注意的问题
T-A 克隆应该时间很简单的事,但知者不难,难者不知啊,还是有很多问题要注意的。T-A 阳性率高,简单易操作,所以在质粒构建过程常常用来选择做为一个亚克隆,既可以用来测序,又有利于进一步酶切,确实很方便。但要注意,首先是 T 载体的选择,尽量避免选择含有目的片段酶切位点的 T 载体,这样会切成多个片段,有时候可能对后面的胶回收会有影响。其次,因为在 PCR 产物是用高保真酶扩增的,所以首先要进行加 A 反应。这时候应购买 T-A 快速克隆平末端加A 试剂盒。进行加 A 时反应体系不要太小,因为太小是酶量加的可能不准确。我开始就是这样,想着节约,说明书写的是 PCR 产物加 15ul,我没舍得,加了 3ul,加A 反应液、酶都相应减量,后面就是转化不成功。后来我 PCR 产物加为 6ul,就成功了,屡试不爽。最后,在连接产物加入感受态细胞后,轻弹混匀,稍微离心一下,不要剧烈的震荡,反正我失败的时候都是力度较大,也不知道有没有关系。
四、菌液 PCR
T-A 克隆成功后,你可以看到很多蓝白斑,一般来说蓝斑要比白斑稍多。不是每个白斑都是你要的东西,下面就是阳性克隆的筛选了。挑白斑到摇菌管过夜摇菌,然后进行菌液 PCR。关于菌液 PCR 引物的问题,有的人用克隆引物,有的人用专门设计的检测引物,两者我都用过,只要检测引物能 P 出来,克隆引物也能 P出来,完全符合,所以一般的时候用克隆引物就可以了,除非运气不好,真好在起始点就出现错配的情况。
五、碱法小提质粒
挑取阳性克隆的菌液,抽提质粒吧。既然是小提,就不要很多菌液,一管就够了,3-5 毫升,开始以为菌液越多越好,其实是错的,首先这点菌液提的质粒够你测序及酶切用了,其次加多了反而不好,因为加的裂解液是相对固定的,多了可能裂解不完全啊。提取质粒有传统的自己配制的酚氯仿抽提,也可以购买试剂盒。我还是倾向于用试剂盒,尤其是像我们这些并不打算在实验方面有所专长的初学者。国产的试剂盒很便宜,如天根的质粒小提试剂盒,一次大约也就 4 块钱,足够用的了。而用酚氯仿抽提吧,很容易出现质粒不纯的情况,在测序时无法测出信号。当然,这也可能跟个人有关,师姐们都建议后者,在他们看来,简单实用,价格更便宜。如何选择,就看自己的了。
六、酶切胶回收
测序结果正确的话那就恭喜你了,至少你已经拿到了目的片段,下面就酶切胶回收吧。将目的片段及载体进行酶切,并行酶失活,然后电泳胶回收。我回收过几次,但回收效率不高,也不知是什么原因。跑胶时条带很亮,酶切也很充分,但一回收就没了,尤其是目的片段常常都看不到。很郁闷,这时候我多做了两个酶切回收体系,同时进行胶回收,加大量回收后跑电泳总算能看到目的片段了,虽说不亮,但总算有。个人认为,将 50ul 的酶切体系不要放在一个孔里面跑胶而是分成两到三个孔跑胶然后回收可能会增加回收的效率。
七、酶切产物连接
载体与目的片段的摩尔比常常为 1:3-10,但酶切回收后的产物测浓度常常测不出来。怎么去按这个比例进行连接反应呢。回收后将目的片段和载体同时跑胶比较亮度,根据亮度的比较再结合分子量来确定两者的体积比。如亮度比为 5:1,两者的分子量比 10:1,体积比大约为 1:3. 不要担心产物的浓度低会影响连接,其实连接只要那么一点点就够了。我的阳性对照加的质粒只有 0.1ng,结果菌落长得满满的。
八、挑取阳性克隆送测序
转化成功后挑取阳性克隆的菌液进行质粒小提,同时保存菌种,送测序。
 关注我们
关注我们